L’Università degli Studi Roma Tre è un’università giovane e per giovani, è nata nel 1992 ed è rapidamente cresciuta sia in termini di studenti che di corsi di studio offerti. Sono attivi 13 dipartimenti che offrono corsi di Laurea, Laurea magistrale, Master, Corsi di perfezionamento, Dottorati di ricerca e Scuole di specializzazione
L’Università degli Studi Roma Tre è un’università giovane e per giovani, è nata nel 1992 ed è rapidamente cresciuta sia in termini di studenti che di corsi di studio offerti. Sono attivi 13 dipartimenti che offrono corsi di Laurea, Laurea magistrale, Master, Corsi di perfezionamento, Dottorati di ricerca e Scuole di specializzazione
Salute: Scoperto l’impatto della mutazione della sequenza R138Q del gene FMR1 nella Sindrome dell'X-fragile
Link identifier archive #link-archive-thumb-soap-81930
COMUNICATO STAMPA Salute: Scoperto l’impatto della mutazione della sequenza R138Q del gene FMR1 nella Sindrome dell'X-fragile Un team internazionale di ricercatori, tra cui l’Università degli Studi Roma Tre, ha permesso di comprendere meglio i meccanismi alla base della sindrome dell'X-fragile, una malattia rara caratterizzata da disabilità intellettiva e autismo. Lo studio, da poco pubblicato sulla prestigiosa rivista Nature Communications, ha individuatocambiamenti strutturali a livello delle sinapsi, deputate alla comunicazione tra i neuroni, causati da mutazioni del gene FMR1, tra cui nella sequenza R138Q
Roma, 19 Marzo 2021 - La formazione di sinapsi funzionali nel cervello è fondamentale per stabilire una comunicazione e una plasticità neuronali efficienti, che sono alla base dei processi cognitivi e sociali. Tutto ciò non avviene nella sindrome dell’X-fragile, dove si riscontrano cambiamenti strutturali a livello delle sinapsi, deputate alla comunicazione tra i neuroni, causati da mutazioni del gene FMR1, tra cui quella nella sequenza R138Q.
Sono questi i risultati di una ricerca che ha visto il coinvolgimento di un ampio network di ricercatori italiani e francesi: il gruppo di ricerca della prof.ssa Viviana Trezza del Dipartimento di Scienze dell'Università degli Studi Roma Tre (Italia); i gruppi di ricerca del dr. Stéphane Martin e della dott.ssa Barbara Bardoni dell'Institute of Molecular and Cellular PharmacologyIPMC - Université Côte d'Azur/CNRS (Francia); il gruppo della prof.ssa Maura Francolini del Dipartimento di Biotecnologie Mediche e Medicina traslazionale dell’Università di Milano e il gruppo del dr. Yann Humeau dell’Università di Bordeaux.
I risultati dello studio sono stati pubblicati sulla prestigiosa rivista Nature Communications in un articolo dal titolo Missense mutation of Fmr1 results in impaired AMPAR-mediated plasticity and socio-cognitive deficits in mice – URL: Link identifier #identifier__21417-1https://doi.org/10.1038/s41467-021-21820-1. La sindrome dell’X-fragile è la forma più comune di disabilità intellettiva ereditaria dopo la sindrome di Down ed è la principale causa monogenica nota di autismo. La malattia è causata da mutazioni del gene FMR1 situato sul cromosoma X. Tale gene contiene le istruzioni per produrre la proteina FMRP (Fragile X Mental Retardation Protein), che ha un ruolo cruciale nello sviluppo del cervello. Le mutazioni del gene portano alla mancata espressione della proteina FMRP, determinando la comparsa di importanti disturbi dello sviluppo neurologico e altri deficit che caratterizzano la sindrome dell'X-fragile. In casi più rari, le cosiddette mutazioni missenso nel gene FMR1 causano la sostituzione di un amminoacido con un altro nella proteina FMRP, portando ad anomalie funzionali della proteina e, di conseguenza, alla malattia.
Grazie ad un approccio multidisciplinare, il team internazionale di ricercatori ha rivelato l'impatto funzionale della mutazione del gene FMR1 nella sequenza R138Q. La mutazione R138Q è di particolare interesse poiché è stata identificata in alcuni individui che presentano i sintomi tipici della sindrome dell’X-fragile.
“Lo studio, effettuato su topi che esprimevano la mutazione del gene FMR1 – spiega la dott.ssa Alessandra Folci, prima autrice dell’articolo - mostra che nell'ippocampo di tali topi, un’area cerebrale cruciale per la memoria e l’apprendimento, i neuroni presentano cambiamenti strutturali a livello delle sinapsi, deputate alla comunicazione tra i neuroni. Utilizzando esperimenti di biochimica, microscopia ad alta risoluzione, elettrofisiologia e una batteria di test comportamentali, questo lavoro dimostra anche che i topi che hanno questa specifica mutazione presentano anomalie della trasmissione neuronale e deficit a livello sociale e cognitivo caratteristici della sindrome dell’X-fragile”.
Grazie ai risultati di questo studio, inoltre, la dott.ssa Alessandra Folci è risultata vincitrice di un prestigioso finanziamento Marie Skłodowska-Curie, che le ha permesso di ritornare in Italia, presso l’Istituto Clinico Humanitas di Rozzano, dopo aver trascorso un prolungato periodo di ricerca all’estero nel gruppo del dr. Martin, in Francia.
“Questa ricerca – spiega la prof.ssa Viviana Trezza, docente di Farmacologia del Dipartimento di Scienze dell’Università degli Studi Roma Tre e coordinatrice degli esperimenti comportamentali dello studio - evidenzia l'importanza di studiare le mutazioni missenso al fine di comprendere meglio i meccanismi che vengono alterati e che sono alla base della patologia umana. Questo modello animale ha rivelato l’impatto della mutazione del gene FMR1 nella sequenza R138Q ed è un utile strumento di indagine per valutare l'efficacia di nuovi possibili trattamenti farmacologici per i pazienti affetti da questa particolare mutazione”.
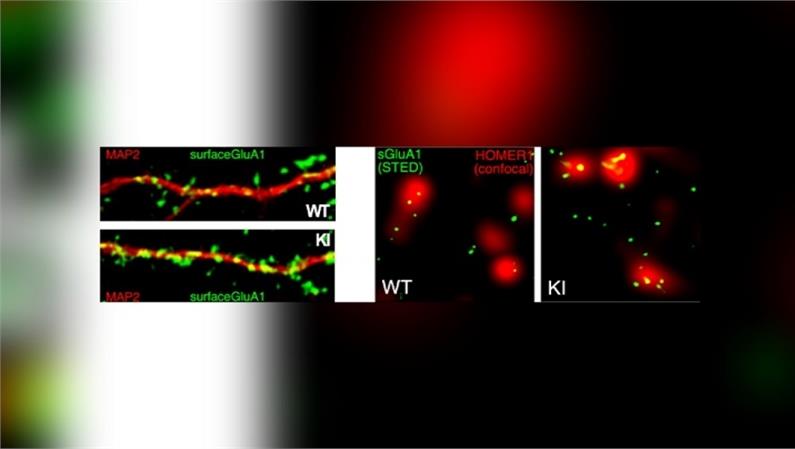
Didascalia immagine: Nel pannello a sinistra abbiamo colorato in rosso il ramo di un neurone di ippocampo di topi controllo (WT) e portatori della mutazione R138Q (KI). In verde, invece, abbiamo colorato il recettore AMPA, una proteina importante per la comunicazione da neurone a neurone. Il pannello a destra è stato ottenuto usando un microscopio a super risoluzione e mostra ancora la proteina AMPA (verde) contenuta dentro le sinapsi (rosse), che sono le strutture attraverso cui i neuroni comunicano tra loro. Le nostre analisi hanno dimostrato che i topi portatori della mutazione R138Q hanno una maggior espressione di AMPA rispetto ai controlli.
CONTATTI UFFICIO STAMPA Dipartimento di Scienze Università degli Studi Roma Tre Francesca Vitalini | Link identifier #identifier__10847-2francesca.vitalini@uniroma3.it | 339 339 0878
Università degli Studi Roma Tre Alessia del Noce | Link identifier #identifier__33300-3alessia.delnoce@uniroma3.it | 339 5304817
Questo sito utilizza un cookie tecnico per consentire la corretta navigazione. Confermando accetti il suo utilizzo. Se vuoi saperne di più e leggere come disabilitarne l'uso, consulta l'informativa estesa. ENGAccettaInformativa completa